Hypertrophic cardiomyopathy (HCM) is a clinical condition, but its name has been subjected to frequent changes over the years, largely because of its morphological and functional heterogeneity, which leads the clinician who is focused on its study to have difficulty in understanding how to diagnose it and when and how to treat it. Regarding its name, it has been called in more than 75 different ways, and it has being classified with difficulty through echocardiography for more than 40 years. Today, it is necessary to understand that the diverse phenotypic behavior, as well as the evolutionary stages of the disease, must be approached in a practical and effective way, so that it easier to understand its clinical behavior and prognosis, as well as the therapeutic needs in each particular case. We review the aspects related to the name of the condition and propose a new that could provide the clinical and surgical cardiologist a better understanding of Hypertrophic cardiomyopathy (HCM) in its various morphological and functional aspects.

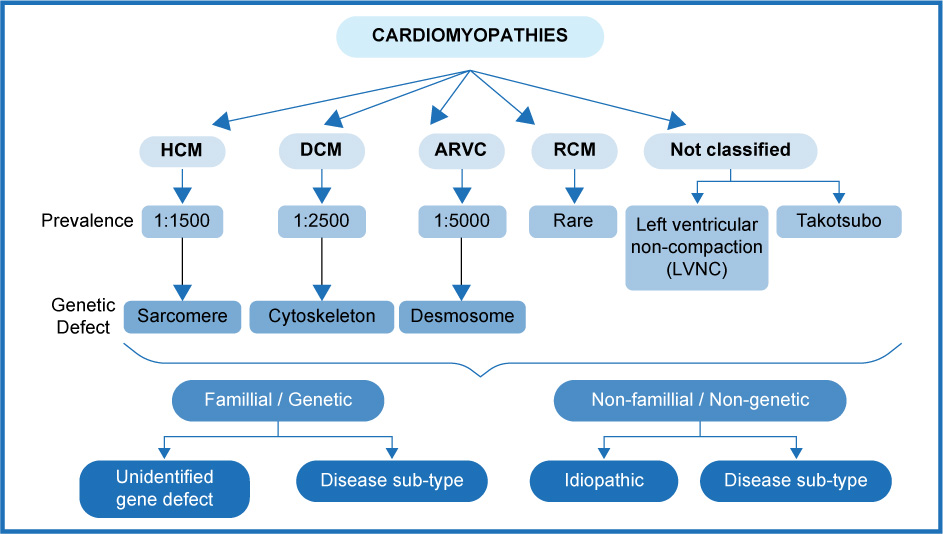
The nomenclature and Classification of Cardiomyopathies have had constant changes since 1961, when Goodwin et al. published what may be considered the first descriptive article of the clinical aspects of Cardiomyopathies, which from the pathophysiological point of view were divided into congestive, constrictive and / or obliterative and obstructive. As early as 1957, Bridgen had coined the term "Cardiomyopathy" when referring to non-coronary heart disease, restricting the term "myocarditis" only to encompass patients with inflammatory disease of infectious etiology. In 1980, the World Health Organization (WHO) defined cardiomyopathies as a disease of the cardiac muscle whose cause was unknown and in 1995 the WHO together with the International Society and Federation of Cardiology (ISFC) developed the joint work (“Task Force”) for the Definition and Classification of Cardiomyopathies (CM), and expanded the classification to include all the diseases that affected the cardiac muscle (myocardial diseases associated with myocardial dysfunction) taking into account both, the etiology and the dominant pathophysiological pattern, and they included in it: dilated cardiomyopathy, hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy and for the first time they added arrhythmogenic right ventricular cardiomyopathy, which had been described in 1982. It was until 2006, when Maron et al., endorsed by the American Heart Association (AHA), proposed a classification and sub-classification, where primary cardiomyopathy (which affects only or primarily the heart muscle) could be named according to its origin as genetic, mixed and acquired, providing a description of each one. Hypertrophic cardiomyopathy was defined as a genetic primary CM, and its origin was attributed to various mutations of sarcomeric proteins encoded by 11 genes. In 2011, The American College of Cardiology (ACC), together with the AHA, endorsed their classification published 5 years before, with the participation of Maron himself. Two years later, the MOGE(S) classification for Cardiomyopathies appeared, and it included a phenogenotypic nomenclature system endorsed by the World Heart Federation (WHF), and which attempted to use a descriptive system of nomenclature and notation applied to diagnosis in the clinical practice (like the one used in oncological diseases) based on the knowledge of the genetics and pathophysiology of each cardiomyopathy, and they also placed the acronym S to refer to the functional class of the described condition. Despite everything, in the European Guidelines for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy published in 2014, little value was given to the MOGE(S) Classification, and they adopted the recommendations of the ESC itself. They defined them with morphological and functional criteria with the family / genetic and non-family / non-genetic subtypes regardless of the presence of extra-cardiac disease. Hypertrophic Cardiomyopathy was defined by the presence of an increase in the thickness of the left ventricular wall which cannot be explained by abnormal overload conditions (Figure 1).

The first description of Hypertrophic Cardiomyopathy was made by Schmincke et al. in 1907 in autopsy results.In 1957, Brock called it aortic subvalvular stenosis and in the same year, Donald Teare published his descriptive article of nine autopsy cases, which he had described as asymmetric cardiac hypertrophy in young adults; seven had suffered sudden death. He described the disorganization of the myocardial fibers for the first time and he even classified it as a muscular hamartoma.
In 1962 in Mexico, Fishleder et al. described the clinical, auscultatory and exploratory findings in patients that they considered had a dynamic subaortic stenosis.In 1964, Braunwald, Morrow et al. delineated the condition that they called idiopathic hypertrophic subaortic stenosis, based on the analyzes of 64 patients, which included Morrow himself, who described the well-known Morrow Myectomy, and who suffered the disease. 14 That same year, Cohen et al. called it Obstructive Hypertrophic Cardiomyopathy for the first time. It should be mentioned here that, to date, the disease has been called in more than 75 different ways, which has generated greater confusion in its understanding.
Today we know that Hypertrophic Cardiomyopathy is a hereditary disease transmitted in an autosomal dominant manner, characterized by an inexplicable hypertrophy of the myocardium, which has an estimated phenotypic prevalence of 1 in 500 (2 per 1,000) in an urban adult population with a similar prevalence in different continents and countries and a possible genotypic prevalence of 1 in 200 (5 per 1,000) which places it probably as the most common of the genetic heart diseases.To support this last hypothesis, there are several avenues of evidence, such as the fact that pathogenic sarcomere genes are more common in the general population than previously thought, as well as the fact that the genetic tests with a greater capacity that are carried out today have defined a new subgroup of patients, who don’t have clinical expression or left ventricular hypertrophy (positive genotype, negative phenotype); and on the other hand, thanks to the image technology currently used, such as cardiac magnetic resonance imaging (MRI), the recognition of some Hypertrophic Cardiomyopathy phenotypes that are difficult to delineate by means of 2D echocardiography, is more feasible. In addition, it should be remembered that previous prevalence studies did not consider the hereditary nature of the disease, whereas now it is common to perform a family detection. So far, a little more than 11 genes, which may encode more than 1500 mutations in cardiac sarcomere proteins have been identified. Around 60% of young and adult patients with Hypertrophic Cardiomyopathy have one of these mutations and 5-10% are caused by another type of metabolic or neuromuscular genetic alteration. Its origin is still unknown in 25-30% of the patients.

The WHO determined to use the term hypertrophic cardiomyopathy as it is considered the most
accurate term to describe this primary hypertrophy that can occur with or without dynamic left
ventricular outflow tract obstruction (LVOTO). Therefore, today it has been classified from the
hemodynamic point of view, into two large groups:
The Obstructive Form:
Hypertrophic obstructive cardiomyopathy (HOCM), which shows an obstruction to the LVOT which can be present at rest (persistent), can be latent (absent at rest but provoked by some maneuver or effort) or labile (variable). The first two (resting and latent) represent 70% of the cases. The two most common forms of obstruction are the one that is classified as subaortic (which is more frequent and is related to an LVOT obstruction, and the mid-ventricular, in which the obstruction, in case it is present, is precisely mid-ventricular). The LVOT obstruction may be conditioned either by the systolic anterior motion (SAM) of the anterior leaflet or posterior leaflet of the mitral valve, or it may be conditioned by alterations or malposition of the papillary muscle and / or the chordae tendineae (tendinous cords), which by a drag (Venturi effect) causes an incomplete support on the septum, which also frequently favors the appearance of insufficiency of the mitral valve. The mid-ventricular obstruction can be caused by an abnormal insertion of the anterior papillary muscle or by excessive mid-ventricular hypertrophy or by the papillary muscle itself, with a pathological alignment. Both may coexist in the same patient. There may also exist an obstruction in the right ventricular outflow tract, and it is seldom considered important.
In general, patients with the obstructive type of the disease are more symptomatic and have a worse prognosis. LVOT obstruction is closely related to higher mortality caused by obstruction or by sudden death. In the asymptomatic or slightly symptomatic patient, the magnitude of the obstruction loses predictive value for mortality and on the contrary, in the highly symptomatic patient, mortality is higher regardless of the degree of the obstruction.
The non-Obstructive Form:
The non-obstructive hypertrophic cardiomyopathy (nHCM) is characterized by disproportionate hypertrophy of the ventricular wall, but it is not possible to demonstrate an obstruction at any level at rest, and it may not be caused by maneuvers such as the Valsalva maneuver or exercise. These patients generally have a more benign and less symptomatic course than patients with the obstructive form.

In 2012, Olivotto I. et al.described 4 Hypertrophic Cardiomyopathy progression patterns, which they classified into Stages I to IV, which are an excellent description of what the natural history of the disease usually is. They allow a clear understanding of the pathophysiological process of the condition. These patterns are:
- STAGE I (HCM in the nonhypertrophic phase), it is characterized by the absence of hypertrophy of the left ventricle (LV) in the context of the subject who has the genotype, usually detected in a systematic family study of a patient who has the disease. The phenotypic manifestation in the patient with the genetic alteration usually manifests itself during the 2nd decade of his life and it may even appear in later stages. An important point to consider is that we are not referring to the patient who has the genotype and who has a negative phenotype (genotype + / phenotype -) for the rest of his life. In this stage. the patient usually has some electrocardiographic manifestations, diastolic dysfunction, and slight dilation of the left atrium, as well as abnormalities of the subvalvular apparatus in the echocardiogram (ECHO).
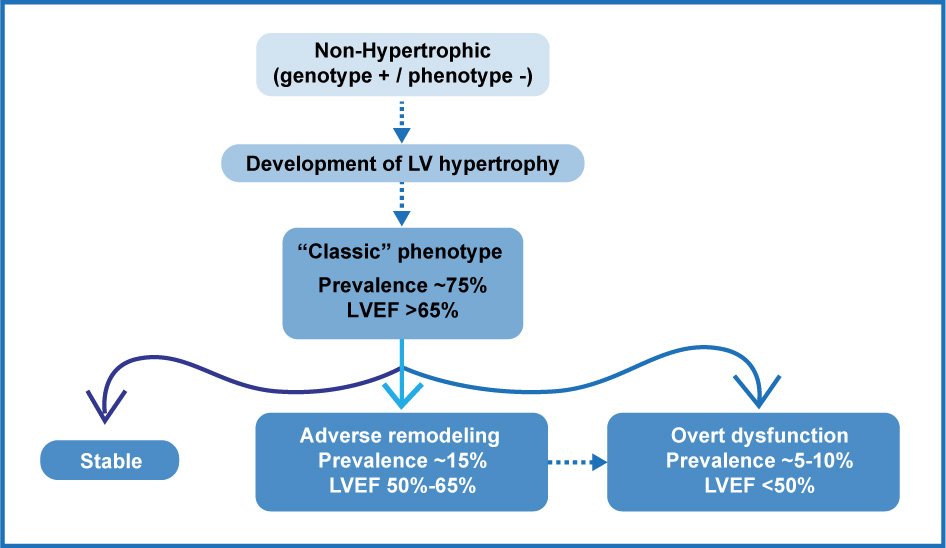
- STAGE II (Classic HCM phenotypically) it refers to the stage in which phenotypic expression becomes manifest, with a hyperdynamic LV, with a LV expulsion fraction (LVEF)> 65%, in the absence of fibrosis. 75% of the patients will enter into this stage, depending on the penetrance of the causal genetic mutation. Typically, regional and asymmetric hypertrophy is observed, most often involving the anterior basal septum and the basal portion of the anterior free wall of the LV. The anatomic patterns of hypertrophy may be multiple (see below). Microscopically, myocardial fiber derangement, microvascular remodeling and incipient fibrosis appear. In this stage, the LV is usually small, hypercontractile, and with an LVEF usually greater than 65%. The fibrosis determined by MRI, and late gadolinium enhancement (LGE) is usually 2% in a (low) average. This stage usually lasts for years or decades.
- STAGE III (adverse remodeling), this stage of the disease has been called by some as "adverse remodeling" and it is defined by the presence of unfavorable structural modifications superimposed on the classic pattern of Hypertrophic cardiomyopathy, a consequence of an increase in myocardial fibrosis and the consequent depression of the systolic function. It is important to mention that apparently this is not an expected average behavior in a patient with Hypertrophic cardiomyopathy as time goes by, but rather a selective pathway followed by approximately 15-20% of the patients, of which a small proportion, will progress to a severe dysfunction and heart failure. It has also been observed that this adverse remodeling is more prevalent in complex genotypes (patients with multiple mutations), which could reflect a profound disorder of sarcomere mechanics and cardiomyocyte energetics. The development of both, the extension and the time of evolution of this adverse remodeling are very heterogeneous, and it is possible to observe it in any age (including childhood and adolescence) and it will progress to a severe dysfunction and heart failure in some cases in a short time or, as in the majority of the cases, it occurs gradually evolving over years or decades. Of course, these patients will go through intermediate stages of progression that go from the hypercontractility phase to the reduced left ventricular systolic function (LVEF) phase, which includes a moderate reduction in LVEF, moderate to significant diastolic dysfunction and a progressive dilation of the left atrium, an increase in the percentage of fibrosis measured by RT, severe microvascular dysfunction, progressive thinning of the LV walls, appearance of atrial fibrillation, spontaneous reduction or loss of LVOT obstruction, and the appearance of apical aneurysm. Not all of these, but some coexist in the same patient, and this entails the presence of a poor evolution and progression towards adverse remodeling.
In general, this remodeling model behaves in a variable way with the appearance of moderate to large areas of intramyocardial fibrosis that can be confluent or in patches, which replace significant portions of the LV, usually from the middle portions of the wall that tend to radiate towards the subendocardium (although they can be transmural). This causes the LV wall to be thinner and it creates cicatrization or repair processes. Finally, it seems that everything is a consequence of microvascular ischemia, cardiomyocyte energy depletion and apoptosis that leads to a progressive loss of myocytes that are replaced by fibrous tissue.
- STAGE IV (excessive dysfunction), although it is rare, it is characterized by significant LVEF deterioration (<50%), significant fibrosis and LV dilation with clinical manifestations of heart failure. Although it may be considered as an “end-stage” disease, some people have called it a “burned out” phase and it corresponds to ~5% of patients. Maron BJ et al.had already defined this stage, when in 1998, they described the progression of some patients with Hypertrophic Cardiomyopathy to systolic dysfunction, in whom the wall thickness was reduced by approximately 25% in 5-6 years (from 20 to 15 mm in average) at a rate of 1 to 2 mm per year, and the LV cavity measured in diastole, dilated 20% in this period (from 45 to 55 mm in average) at a rate of 1 to 1.5 mm per year. (Fig. 2).
A very important aspect is that in this stage, they cataloged two types of patients. The first type was defined as "dilated hypokinetic" characterized by an increased volume and remodeling with LV sphericity, and sometimes it was difficult to differentiate it from cases with dilated cardiomyopathy of idiopathic type. Usually at this stage the LVOT obstruction has disappeared. It is common to find pulmonary arterial hypertension, biventricular dilation, and mitral regurgitation due to dilation of the valve ring (annulus). The second form was called “hypokinetic-restrictive” characterized by a small and rigid LV cavity with significant diastolic dysfunction that simulates restrictive cardiomyopathy; in these patients, systolic function was slightly compromised, and it was possible to distinguish some degree of asymmetry in the thickness of the LV wall. It is now known that this last form is related to some specific type of mutations such as TPM1, MYL3 and MYL2. Although Olivotto et al.23 did not define it this way, Melacini P. et al. described a third pattern in terminal patients, characterized by progressive heart failure because of “persistence of LVOT obstruction”, which eventually conditions dilation and systolic dysfunction of the LV, without the total disappearance of the obstruction. These patients are represented by older individuals (50 ± 14 years at diagnosis). The onset of symptoms after diagnosis is also short (4 ± 6 years) although once started, they rapidly evolve to an advanced functional class (1 ± 2 years).

In Hypertrophic Cardiomyopathy, the distribution of LV hypertrophy is characteristically asymmetric and particularly heterogeneous, it can encompass a wide range of wall thickening patterns, from extensive and diffuse to mild and segmental, and without a single morphological expression considered typical or classical. However, for a better clinical understanding, there has been an effort to subclassify these patterns in such a way that it is possible to understand which could be the dominant hypertrophic point, and thus expect to a certain degree, a predictable clinical evolution. In spite of their diversity, the LV hypertrophy patterns are generally not extensive and usually involve <50% of its wall in half of the patients, and their extension is less in a significant minority.
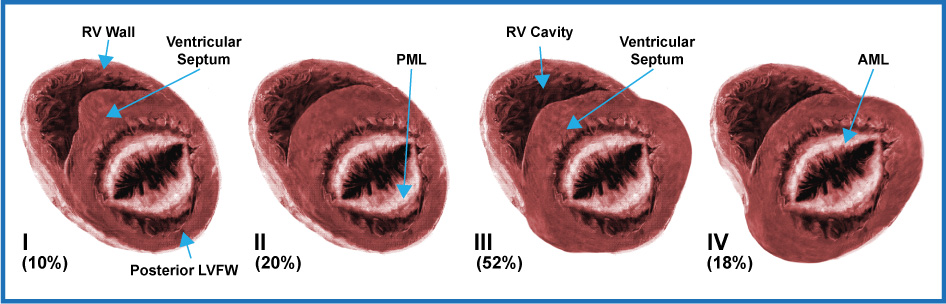
Although the phenotypic characteristics were initially described by autopsy findings and later by 2D ECHO analysis, the first formal attempt to classify Hypertrophic Cardiomyopathy according to its anatomical characteristics was made by Maron BJ et al. in 1981, through the analysis of 125 patients with the disease, who underwent a 2D ECHO. They defined 4 dominant LV hypertrophy distribution patterns, which were called Type I - IV based on their location:
- Type I (10% of the cases), it is characterized by having hypertrophy confined only to the anterior portion of the LV septum,
- Type II (20% of the cases), it corresponds to patients with hypertrophy of the entire ventricular septum (anterior and posterior segment) but not to the LV free wall,
- Type III (the most frequent), it corresponds to hypertrophy of basal portions of both the septum and the LV free wall (52% of the patients), and of these, a small proportion (25 in 65 patients) had a predominance of hypertrophy in the apical regions; and finally,
- Type IV (18% of the patients), with involvement of other LV regions and without showing hypertrophy of the anterior basal portion of the ventricular septum.
(Fig. 3).
They confirmed that previous studies performed by M-mode echocardiography significantly underestimated (up to 5 mm) the maximum thickness of the septal or free LV wall. That same year, Yamaguchi H et al. published the ventriculographic and echocardiographic findings of 30 Japanese patients with apical Hypertrophic Cardiomyopathy. They had a marked apical obliteration together with "giant" negative T waves in the electrocardiogram with high voltage QRS and a thickness of the apex wall > 15 mm. In Maron's Description, patients with apical HCM in Type III and IV had been included, but it did not include concentric hypertrophy. Maron himself in 1985, described 2 patients with posterobasal LV free wall hypertrophy with evidence of obstruction. This morphological type was not included in the classification. One year later (1986) Candel-Riera et al. added two types to the classification proposed by Maron:
- Type V which corresponded to concentric hypertrophy, and
- Type VI which corresponded to the apical hypertrophy described by Yamaguchi.
In the year 2000, Romero-Farina et al. compared Maron's echocardiographic classification with myocardial morphology analyzed by myocardial tomography scan (SPET). Although their power of spatial resolution was lower than the ECHO and the other techniques such as MRI, they considered that allowing the distribution of hypertrophy to be observed, facilitated the classification of patients into different morphological types. They analyzed 119 patients and found that only in 64% of them, the ECHO was able to facilitate the measurement of all the LV segments, and with the SPET they obtained a more complete visualization of all the segments in the short axis, so although the spatial resolution did not allow them to accurately measure the thickness of the walls, it made it possible to establish which morphological type each case corresponded to. They confirmed that the most frequent type was Maron's type III (74% of the cases), but in 25% of the cases the ECHO made a mistake in defining the classification fundamentally in the same type. They also found that 4 patients could not be classified within type I - VI (they had septal and inferior hypertrophy).

In 2007, Maron et al. described the involvement of the right ventricle (RV) when studying 46 patients with Hypertrophic Cardiomyopathy who underwent MRI and they compared them with 22 healthy individuals carefully analyzing the thickness of the wall of both ventricles. They found that the maximum RV wall thickness was greater in Hypertrophic Cardiomyopathy patients compared to the control group (7 ± 2 vs 5 ± 1 mm, p <0.001), including 15 (33%) with a maximum thickness ≥ 8 mm, and four (9%) with extreme hypertrophy (≥ 10 mm). They demonstrated a close correlation between the degree of LV hypertrophy and RV hypertrophy. The greater the hypertrophy of the former, the greater the hypertrophy of the latter (R2 = 0.4, p <0.001). The presence of RV hypertrophy was never included in Maron's classification. In the same way, in 2010, Keeling et al. published the case of a patient with Hypertrophic Cardiomyopathy in whom they detected RV hypertrophy through MRI but who also had apical fibrosis and fibrosis of the free wall of this ventricle, which had been detected by RT. It is now known that in addition to RV wall hypertrophy, it is common to find a prominence of some RV structures, such as the supraventricular crest (Wolf's spur) that is located between the pulmonary and right atrioventricular orifices and that can lead to confusion when measuring right septal thickness. The most important portion to evaluate is the right septal basal portion and the basal portion of the RV free wall, which can sometimes condition RV outflow obstruction and it requires surgical treatment.

Apical Hypertrophic Cardiomyopathy, initially described by Sakamoto et al., and later discussed masterfully by Yamaguchi et al. is a phenotypic variant apparently modulated by environmental and genetic factors, but in general its diagnosis is a challenge when only ECO 2D is used. Although the apical LV involvement is a sine qua non component in the apical form of HCM, some mid-ventricular segments may also be hypertrophied (this is one of the mixed morphological subtypes), in which mid-ventricular obstruction with obliteration may occur of the LV cavity. These patients may present an apical aneurysm (AA), which is usually another aspect that is difficult to diagnose, and which is usually underdiagnosed and undervalued. In patients with apical Hypertrophic Cardiomyopathy, the presence of AA can be detected in 2% to 28% of patients (there is a higher incidence in patients with obstructive mid-ventricular hypertrophy). This anomaly corresponds to a small segment of the LV apex, with a thin, dyskinetic or akinetic wall, with a relatively narrow neck that communicates with the basal or medial portions of the LV. Patients with AA associated with mid-ventricular hypertrophy represent an important subgroup of patients who, in addition to being usually underdiagnosed if MRI is not used in their analysis, have special prognostic and therapeutic implications. MRI has demonstrated that AA is usually made up of fibrous tissue and that RT can be infiltrated into the adjacent septum and free wall, representing a “nest” which fosters ventricular tachyarrhythmias and thus increases the risk of sudden death. In 2008, Maron et al. demonstrated that AA patients had a higher risk of progressing to heart failure, having sudden death, generating systemic embolism and having apical thrombi. The average risk of adverse clinical consequences in these patients was 10.5% per year, which is considerably higher than the amount reported in the totality of the patients with Hypertrophic Cardiomyopathy. On Holter monitoring, more than 40% of these patients had monomorphic nonsustained ventricular tachycardia, which is considered high risk for sudden death in patients with HCM. AA may also be present in patients with pure apical H Hypertrophic Cardiomyopathy.(Figure 4)
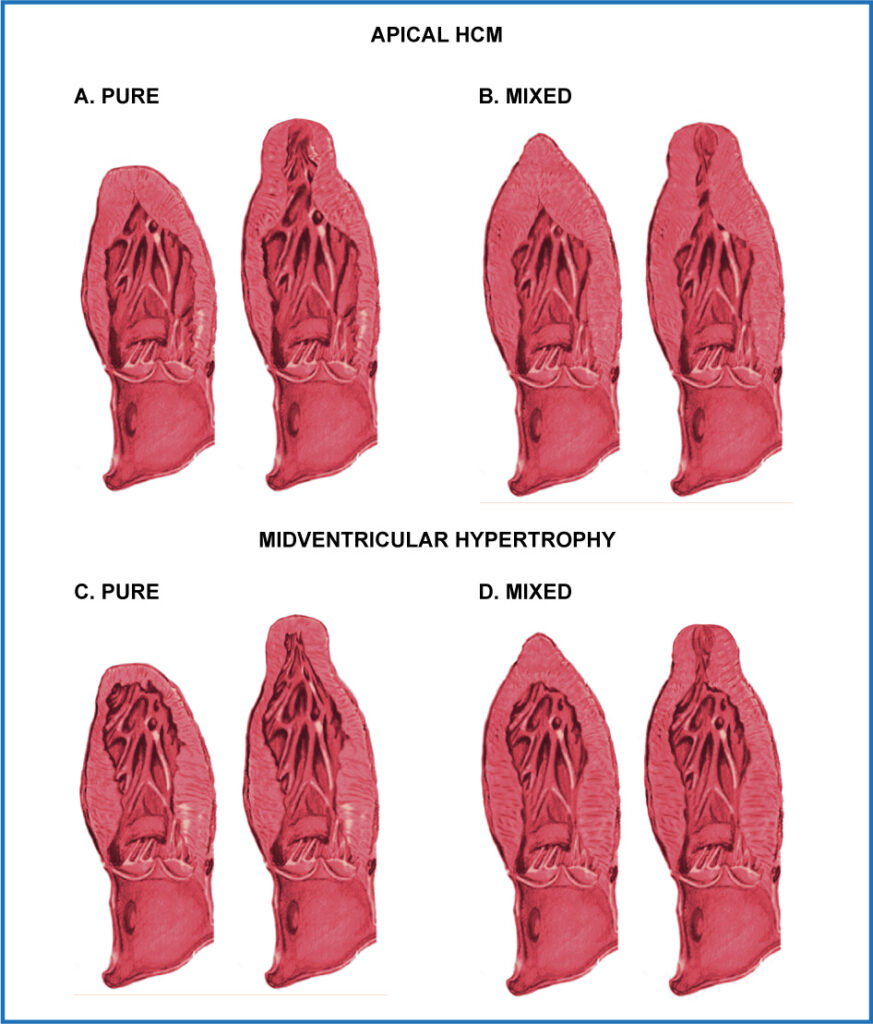
Figure 4. Classic phenotypes and subtypes of mid-ventricular and apical hypertrophic cardiomyopathy. A: isolated apical form (Type III), it is the traditional phenotype, limited to the apical segments of the LV, it may have an apical aneurysm (Type IIIa). B: mixed balanced form, in which the contiguous (mid-ventricular), and rarely the basal portions, are also hypertrophied, although apical hypertrophy predominates (Type III). It may or may not have an apical aneurysm. C: phenotype with isolated mid-ventricular hypertrophy, without involvement of the apical segments (Type II). It may or may not have an obstruction and an apical aneurysm. D: predominantly mid-ventricular form, in which this region has a greater hypertrophy (Type II), but the LV apex is also hypertrophied and it has an obstruction. It may have an apical aneurysm (Type IIa-o). (Modified from Jan et al.).
The presence of mid-ventricular obstruction with cavity obliteration is frequent and it is only possible to observe it in apical Hypertrophic Cardiomyopathy of the mixed type (mid-ventricular involvement) and in severe cases. the obliteration of the cavity persists until the end of the diastole, and it is associated with the presence of a paradoxical diastolic flow jet towards AA, which is usually a marker of poor prognosis.

In 2009, Maron et al. characterized the distribution pattern of LV hypertrophy using MRI, seeking to define more precisely the phenotypic expression of these patients. They studied 333 patients who had Hypertrophic Cardiomyopathy with MRI. They considered that since this offered a great spatial resolution and a complete anatomical reconstruction of the LV, it would allow them to evaluate the precise distribution of hypertrophy. It was thus possible to define that the contiguous basal portions of the anterior free wall and the ventricular septum (number 1 in the clock of the short-axis plane), it constituted the regions with a predominance of thickening (77%) that corresponded to the distribution known as Maron Type III.They found that just over half of the patients had areas of hypertrophy distributed over ≥ 50% of the entire LV chamber (diffuse hypertrophy), and that a minority had focal or regional areas of hypertrophy. 12% of the patients had only 1 or 2 hypertrophied LV segments, and they were classified as focal involvement, which were not even enough to show an increase in the calculated total LV mass. They found a moderate involvement that represented between 13% and 49% of the entire LV (3 to 7 segments) in 34% of the patients, and finally diffuse involvement (≥ 8 segments), in other words, > 50% of the entire LV, in 54% of patients. Frequently, the phenotypic expression was non-contiguous segmental hypertrophy, which created abrupt changes in wall thickness (in patches separated by non-hypertrophied regions), which were present in up to 15% of the patients, and which they describe as a lumpy aspect. In this last group, it was common to find combinations of hypertrophy in patches of the basal anterior septum and in the apical anterior wall, as well as the basal anterior septum with the posterior medial septum.
In this study, Maron et al. also demonstrated that by means of RT, it was possible to identify and quantify "in vivo" the presence of myocardial fibrosis which is so common in patients with HCM. They also found that the extent of hypertrophy (number of hypertrophied segments) was related to the presence of LVOT obstruction and to a greater deterioration in their functional class of heart failure. The greater the hypertrophy, the greater the degree of obstruction and the greater the clinical deterioration.
In this study, various hypertrophy patterns were defined as follows:
a) Hypertrophy of the LVseptum preserving the free wall,
b) Focal hypertrophy of the anterior basal portion of the LV septum,
c) LV apex hypertrophy,
d) Segmental hypertrophy predominantly of the anterolateral LV wall with normal anterior septum,
e) Massive asymmetric hypertrophy of the anterior wall of the LV septum, without involvement of the posterior septal portions,
f) Diffuse hypertrophy with involvement of the septum and the anterolateral wall.
Furthermore, they concluded that using 2D ECHO, LV wall thickness was underestimated in some patients, since the epicardial border of the free wall was frequently not adequately visualized. They concluded that MRI is the optimal method to precisely define the morphological characteristics in the patient with Hypertrophic Cardiomyopathy. Since then, no more has been said about Maron's echocardiographic phenotypic classification, which has been forgotten by many people.
Since then, more than an anatomical classification, the concept of phenotype has been used to refer to the morphological and functional aspects of Hypertrophic Cardiomyopathy. The most common morphologies of the septum and the distribution of hypertrophy in the left ventricular wall in patients with Hypertrophic Cardiomyopathy (phenotypic expression) are shown in Figure 5. In fact, this distribution has been used to subdivide the condition into these more commonly identified 4 groups.
As it has already been mentioned, although there are four phenotypic types which are considered as the most frequent in terms of the location of hypertrophy, such as basal sigmoid hypertrophy (with LVOT obstruction), reverse curvature hypertrophy (which usually corresponds to mid-ventricular hypertrophy, and may have mid-ventricular obstruction and / or AA), apical and neutral hypertrophy; in general, some of them may coexist in the same patient, or even have atypical or mixed localization components. A greater extent of left ventricular hypertrophy has been associated with a younger age and a mitral valve with a higher degree of SAM of the valve and outflow obstruction but it has not been shown to be related to the magnitude of the symptoms or to the gender.
Another important point related to the findings of Maron et al. through the analysis of the MRIis that today, it may not be possible to evaluate the patient with Hypertrophic Cardiomyopathy and plan an adequate therapeutic strategy, without a correct non-invasive anatomical diagnosis through a detailed analysis of the MRI, which allows to specify the site or LV portion with greater hypertrophy, as well as the coexistence of morphological alterations of the mitral subvalvular apparatus.


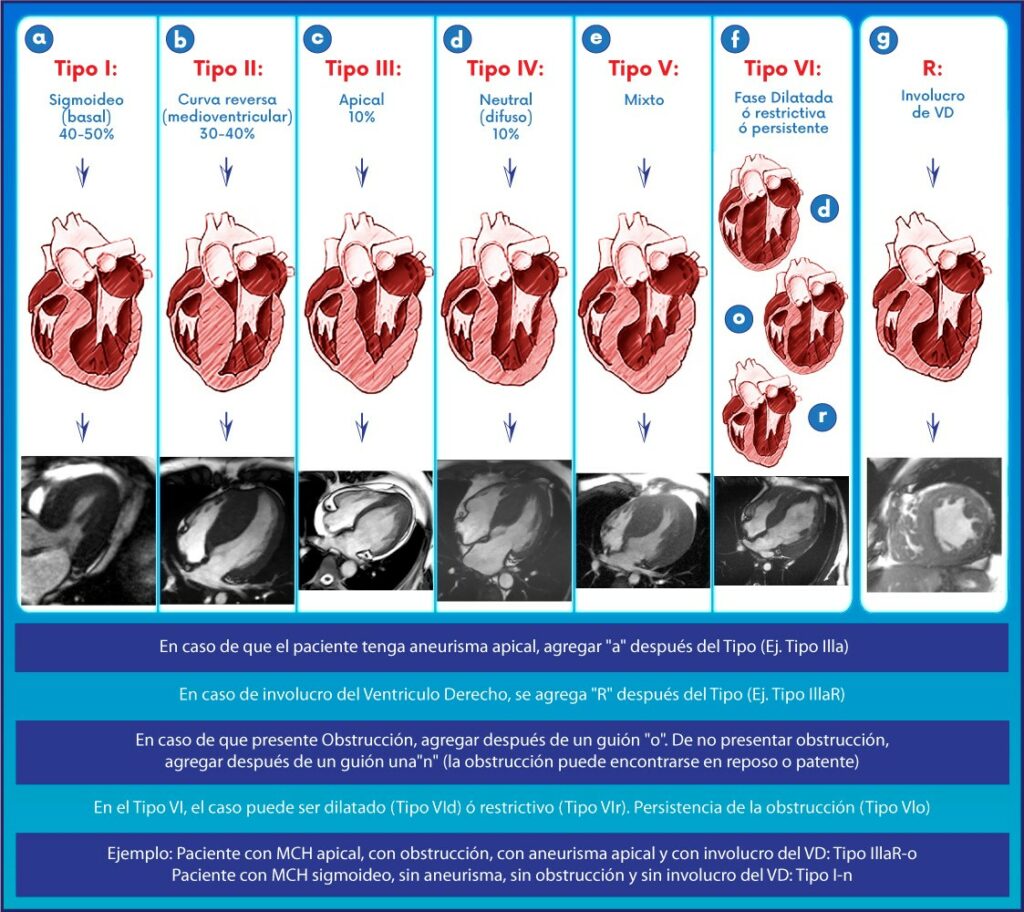
Five forms of phenotypic expression have been identified that are considered the most common in the early stages of Hypertrophic Cardiomyopathy, and one form was identified in late stages of the disease. On the other hand, there are many reports that have made it possible to define the most common morphological manifestations of the disease and now we know that in addition to the LV, the RV may be involved and there may also be an obstruction of the latter's outflow tract. Furthermore, it is known that a late phenotypic manifestation (end stage) of the disease may be an LV dilation phase with significant systolic dysfunction (burned-out). It is known that there is a group of patients with LVOT obstruction (some at rest and others with patent obstruction) and others with mid-ventricular obstruction, and a minority without obstruction at any level. Finally, there are patients who develop LV AA as a serious complication. For this reason, it is increasingly necessary to have an anatomical-functional classification scheme, which allows each patient to be classified into a group that could also have different therapeutic and prognostic implications. For this reason, we are proposing the use of a classification system that facilitates its understanding in a clear and practical way. The proposal is to classify the morphological findings into six Types (Fig.6):
- Type I, would correspond to the “sigmoid” variety with hypertrophy of the basal segments (anterior and / or posterior) of the septum and the free anterior wall, which corresponds to the most frequent (40 to 50% of cases). This is characterized by a prominent basal septal bulge, a concave medial septum, and an ovoid left ventricular cavity (Figure 6a). 40% of type I patients will have a detectable obstruction at rest (persistent) and 30% will have an obstruction provoked by some maneuver (latent), in other words, 70% will have an obstruction. This last group of patients will be those that may be defined as having obstructive Hypertrophic Cardiomyopathy (oHCM).
- Type II, corresponds to patients with mid-ventricular hypertrophy (reverse curvature) and it represents 30 to 40% of all patients. They may also have mid-ventricular obstruction, or this obstruction be absent (≥ 30 mmHg peak instantaneous gradient in 9.4%). (Figure 6b). There is a subgroup of patients in whom mid-ventricular hypertrophy is accompanied by apical hypertrophy, although to a lesser degree. They may or may not have AA, but this type is the one that develops it most frequently (28%).
- Type III, corresponds to patients with apical hypertrophy, which represent approximately 10% of the cases (Figure 6c). In these patients, the hypertrophy may be "purely" apical, or it may be accompanied by moderate mid-ventricular hypertrophy and very rarely by basal hypertrophy (mixed form) and may or may not have AA.
- Type IV, is the group of patients with diffuse, neutral hypertrophy, without obstruction and without a specific segmental pattern. It corresponds to approximately 10% of the cases. Usually, these cases, will not be obstructive (nHCM). (Figure 6d)
- Type V, corresponds to those patients who have mixed patterns; in other words, areas of hypertrophy in patches in various LV sites, and which could be difficult to classify into one of the four types already described, which usually may only be identified by MRI (Figure 6e).
- Type VI, refers to the patient who is in the dilated phase of the disease (burned-out), where it has been clearly defined, either genetically or with previous imaging studies, that he had Hypertrophic Cardiomyopathy of any type and that finally the ventricle dilated and entered a phase of significant systolic dysfunction (Type VId) (Figure 6f). These patients may rarely have a “restrictive” end-stage form (Type VIr) or persist despite dilatation with some degree of LVOT obstruction (Type VIo).
On the other hand, it is proposed in this classification to use additional acronyms, which include three fundamental aspects of the disease:
- First, whether there is an obstruction. If so, the lowercase letter “o” must be used for obstruction, after the corresponding type and a hyphen. If the patient has basal septal hypertrophy, he would be type I, but if he has either a persistent or latent obstruction, he would be type I-o. If he doesn’t have a detectable obstruction, he would be type I-n (adding the letter “n” which means non-obstructive).
- Second, whether there is an apical aneurysm. If so, the lowercase letter “a” meaning aneurysm, would be added. Thus, if the patient has mid-ventricular hypertrophy, he would be type II, but if there is mid-ventricular obstruction and apical aneurysm, he would be Type IIa-o, and if there is no obstruction, but if there is apical aneurysm, he would be Type IIa-n.
- Third, whether there is RV involvement. In the presence of right ventricular hypertrophy, the capital letter “R” would be added at the end of the text (R comes from Right). For example, in the latter case with mid-ventricular hypertrophy, with obstruction and apical aneurysm, if there were RV involvement, it would be Type IIaR-o. (Figure 6).

This proposal seeks to facilitate the morphological and therefore pathophysiological understanding of the patient with Hypertrophic Cardiomyopathy, in such a way that, classifying him within in a particular Type, the therapeutic needs, both pharmacological and surgical, as well as their prognostic implications, can be understood.
The management and treatment of Hypertrophic Cardiomyopathy has evolved since the first descriptions and thanks to the risk stratification models derived from registries and publications regarding a greater number of patients, we now have facilities for the more precise identification of patients at high risk of sudden death and future complications. Through the early recognition and directed management thanks to a morphological and functional classification, it is possible to identify the therapeutic needs and the prognostic implications in each particular case.